
Welcome to the Buettner lab! We are big Swifties over here (don’t listen to Bonnie, we catch her singing along all the time too). The Buettner lab conducts bioinorganic chemistry research, which translated means we study metals in biology. More specifically, we are interested in understanding how nature uses proteins to control metals. We focus on metals like titanium and vanadium because of their abundance, cheap costs, and myriad of uses. Unfortunately, when these metals are around water, they often react in uncontrolled, unwanted ways that leave them in inactive states (this is like iron rusting). Studying the aqueous chemistry of these metals is an exciting opportunity to open up new applications of these metals.
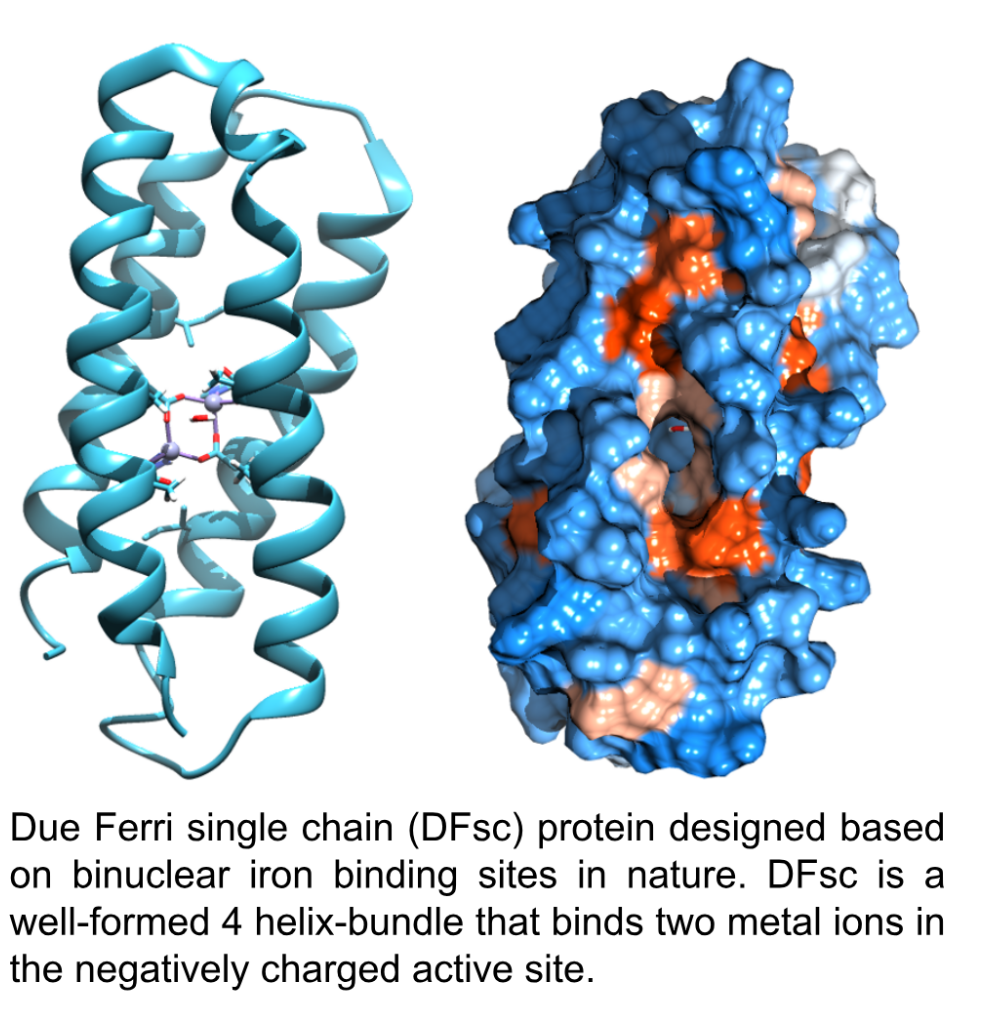
We use a family of computationally-designed proteins called Due Ferri (DF) proteins, which were designed to bind two iron atoms. These proteins are meant to mimic the way nature protects other metals, such as iron, from reacting with water. When we think about iron in an aqueous setting, we think about rust, but nature has built systems to protect iron from reacting with water. For example, Transferrin is a protein in your body that regulates and transports iron in your blood. It protects the iron from reacting with the aqueous environment, allowing it to do some pretty useful chemistry.
Using iron as a model, we design and characterize enzyme active sites that bind to hydrolyse-prone metals and study their reactivity to generate new enzymatic activities.
There are four of us in the lab this summer, and we’re split into binding and activity studies, so follow along as we dig into our research.
A tale as old as Tyrosine
Tyrosine was substituted in for an isoleucine in the active sight for three of our proteins (G14D, DF, G4), creating three new proteins (DY, DFY, G4Y). These substitutions were made because tyrosine has a REPUTATION as a Lewis base, so it should bind well to Ti and V which have strong Lewis acidity. Previous work done in the lab shows the tyrosine substituted proteins bind well to our metals. Ethan and Savarna are studying these six proteins and tyrosine’s effect on metal-protein interactions.
Ethan:

I am a rising junior majoring in chemistry, and this is my second summer in the Buettner lab. I enjoy reading late into the night and going on early morning runs. I focus on six DF protein variants of the Due Ferri single chain proteins. Three of these proteins (DY, DFY and G4Y) have tyrosine, a hard Lewis base, in their active sites. I am looking to see if the substitution of this tyrosine improves the binding interactions of the DF variants and the metals of interest, titanium, vanadium, and zinc. My main goal is to characterize the metal binding of these mini-metalloenzymes. This project not only helps us better understand the role metals play in biology, but also aids in the design and development of new enzymes.
To characterize the binding, I utilize a suite of instruments. One instrument, the inductively coupled plasma optical emission spectroscopy (ICP-OES, I call it the ICP for short), allows me to directly measure the binding stoichiometry of metal: protein. The ICP works by heating up samples to 10,000 K (the temperature of the surface of the sun!) with the help of an argon plasma. Samples are incubated with excess metal and then washed with a buffer to remove any unbound metal. We know that atoms and ions can absorb energy, which moves electrons from the ground state to an excited state. Excited atoms release light at a specific wavelength as they transition to a lower energy level, and the amount of this light released is proportional to the number of atoms in the transition, which allows for information about concentration of metal in the sample to be gathered with the help of a calibration curve. The moles of metal and the moles of protein in the sample are compared to give a binding stoichiometry.


When the ICP isn’t eating up all my time (me and the ICP vibe like that), I am usually at the fluorimeter or the isothermal titration calorimeter (ITC). Proteins are inherently fluorescent because of the tyrosine and tryptophan residues. These tyrosines and tryptophans absorb at 280 nm and the release of a photon is observed at ~350 nm. As we titrate in metal to our proteins, we expect to see an increase in fluorescence, which correlates to an increase in structure. Below is our protein G4 titrated with our control metal zinc. We see an increase in fluorescence as metal is added and the curve starts to flatten out around 2 equivalents, meaning the binding site becomes saturated around 2 equivalents of metal.
Moving forward, I will continue to work with all my instrument friends and look forward to studying different metal and protein combinations. Particularly, I am interested in studying titanium with the fluorimeter to see how it compares to our zinc data. I am also diving deep into isothermal titration calorimetry, which can give us a thermodynamic profile of metal-protein interactions.
Savarna:

Hi! I’m Savarna and I have worked in the Buettner Lab since last semester. I am a rising Sophomore majoring in Chemistry and Health Sciences. I go on morning runs with Ethan and like to binge-watch TV shows in my free time. Running does tire me a ton, but I just SHAKE IT OFF and get to lab 😉 My favorite part of the lab is choosing Taylor Swift songs to play on the lab computer (duh). I must say, I really enjoy doing that (and enjoy doing research too of course). It hasn’t been a CRUEL SUMMER so far, and I can’t wait for more of it!
This summer I am studying the reactivity of our proteins with metals and measuring their ability to cleave DNA and RNA. So far, my focus has been on understanding how the phosphate backbone of DNA and RNA interacts with our proteins, and developing a method to monitor their cleavage.
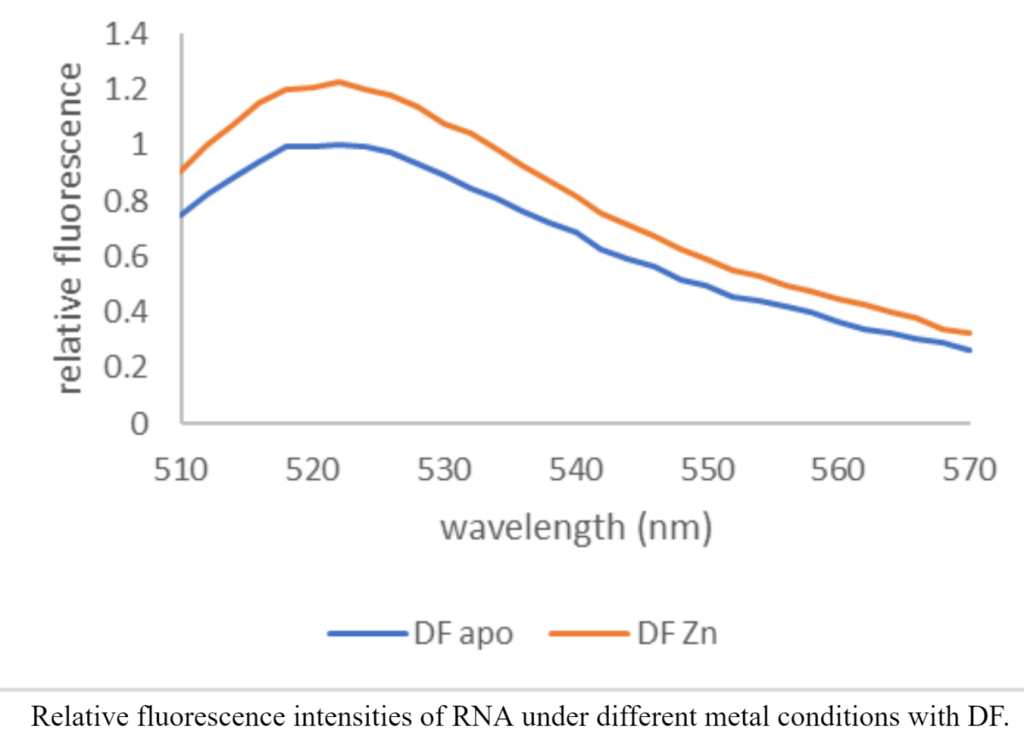
The DNA sequence I use has two ends, with one end being fluorescent, and the other being the quencher. We expect the proteins to cleave the DNA, separating the quencher from the fluorescent end and therefore making the DNA light up! I visually assess the fluorescence under UV light and then use a plate reader to quantitatively assess relative fluorescence. I also do the same with RNA and observe the differences in activity with other protein-metal combinations. It is very exciting to see how different the activity with and without metals is. Just in case you’re wondering (which I’m sure you are), this is what my data looks like! It shows that Zinc-bound protein has higher DNA cleaving activity than just the protein itself.
To understand how the DNA or RNA is actually cleaved, we want to see how phosphate interacts with the binding sites of different proteins and metals. I do this using phosphorus NMR where I look at the shifts in phosphate peaks relative to an internal phosphate control. I expect there to be shifts in the peaks depending on the sample composition and so far I’ve been seeing that! One of the many things I enjoy doing in lab is preparing my NMR samples (and then cleaning the NMR tubes of course). I also love seeing how the NMR instrument works, and am always awed by the amount of data it gives me.
I hope to have a full set of NMR peak shifts by the end of the summer, and am working towards finding out what the shifts tell us about the reactivity of our proteins. That’s a ton of information to look at and I better be READY FOR IT!
All too Arginine
The other side of the protein family has arginine in the binding site to enhance vanadium binding. When V binds to our proteins, it binds as a vanadate ion with a negative charge. The arginine and histidine residues in the active sites make the binding pocket positive, creating a more favorable environment for vanadium binding. Bonnie and Sarah work to determine the effect of arginine in our proteins in order to better understand the binding interactions between our metals and proteins.
Bonnie:

Hi, my name is Bonnie Coley! I’m the one all the way in the back with the pink hoodie! My project is focused on studying the binding aspects of our proteins to specific metals such as vanadium and zinc. I currently work with two techniques: cyclic voltammetry (CV), and fluorescence spectroscopy. By using CV, we can study the redox potentials of vanadium when bound to our proteins by taking advantage of vanadium’s accessible oxidation states. We are looking to see if the redox potential of vanadium changes based on the protein used, which will give us a better understanding of the role arginine plays in metal binding.
Below is a graph of potentials of vanadium alone with no protein present. As the concentration of metal increases, the peak around -200 mV increases in intensity. We can use this control as a reference to compare shifts in the potential with different metals bound to our proteins.

We’re interested in better understanding the binding mechanisms of vanadate. Our proteins have 2 binding sites, so determining how vanadate binds to one, two, or neither of those sites is a main focus in my research. Another way to observe the binding aspects of our proteins is through fluorescence spectroscopy. Our proteins are naturally fluorescent, so by measuring the change in fluorescence upon metal addition, we can learn a lot about metal binding and structural stability.
As I continue my research throughout the summer I hope to gain more knowledge about the science we do in our lab. that I will be able to later apply in my more advanced science courses here at Gettysburg College!
Sarah:
My name is Sarah and I’m a rising sophomore. I am a chemistry major and this is my first summer in the lab. I love spending hours in the gym and ending my nights with a good book. In the lab, I focus on arginine proteins and their reactivity with vanadium to create functional vanadium enzymes. In nature, these enzymes typically function as haloperoxidases. Despite vanadium being a hydrolysis-prone metal, these haloperoxidases can carry out their reactivity in water. With the use of the DFsc protein system, I have been trying to mimic these enzymes and developing assays that enable me to monitor their activity. In doing this, I have been spending a lot of time analyzing color changes with the UV-visible absorbance spectrometer. This instrument measures the light absorbed by my samples at different wavelengths.

However, in order to get more data at once, I’ve been using the plate reader quite a bit. This instrument allows me to collect data on up to 96 samples at once (which is insane!), but that’s a hefty amount of pipetting, so I’ve been sticking with around 12-36 samples at a time. The plate reader enables me to track the absorbance of each sample at a variety of wavelengths and allows me to see and plot what is happening with the enzyme’s kinetics so I can get a better understanding of what is happening with the peroxidases (even allowing me to do this at different temperatures!).
I have been doing two different kinds of assays this summer on the same instrument. The first is a pyrogallol peroxidase assay. My samples consist of the arginine proteins, hydrogen peroxide, vanadium, buffer, and a substrate known as pyrogallol (below).

With this assay specifically, I am looking to see the absorbance of my samples increase as they turn from a clear color to a dark REDdish-yellow or orange. Looking at the plot below, we can see that the absorbance for the arginine proteins (E11R and E44R) with vanadium is higher than both that of the controls and the apo proteins (proteins without metal). This GORGEOUS graph shows what we want to see because for this assay, higher absorbance means more product, and we are looking for the proteins with vanadium to produce the greatest amounts of product.

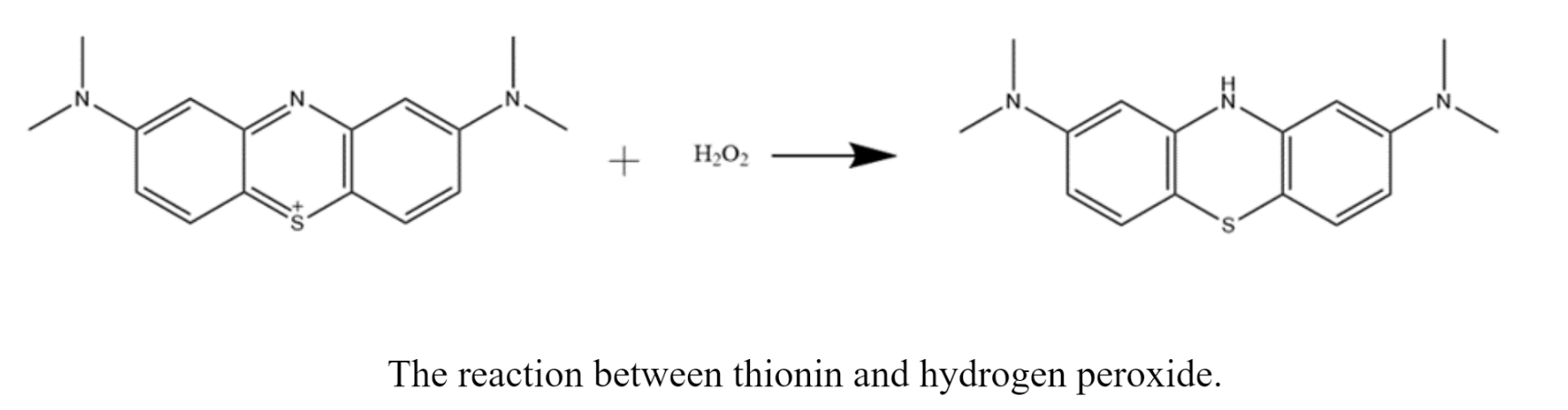
The other assay I do is a thionin peroxidase assay. These samples consist of arginine proteins, hydrogen peroxide, buffer, vanadium, and a deep purple substrate called thionin. With this assay, I am looking to see bleaching of my samples. A colorless product forms as the protein reacts with the purple substrate. This reaction is extremely slow, so I often do a single scan in the UV-vis or plate reader the next day.
The results I am looking for with this assay are the opposite of what we want to see in the pyrogallol assay. In other words, because we are looking for bleaching in these samples, we want the absorbance to decrease. So, when comparing the absorbances of these samples, we want the absorbance of the proteins with vanadium to be lower than the controls and the apo protein samples. I haven’t quite gotten results that show this yet, but SO IT GOES.
I am keeping my hopes up as I change the assay conditions that we will find the sweet spot! At the very least, THIS IS ME TRYING.
Thanks for following alone! Come by and sing along with us some time.



